
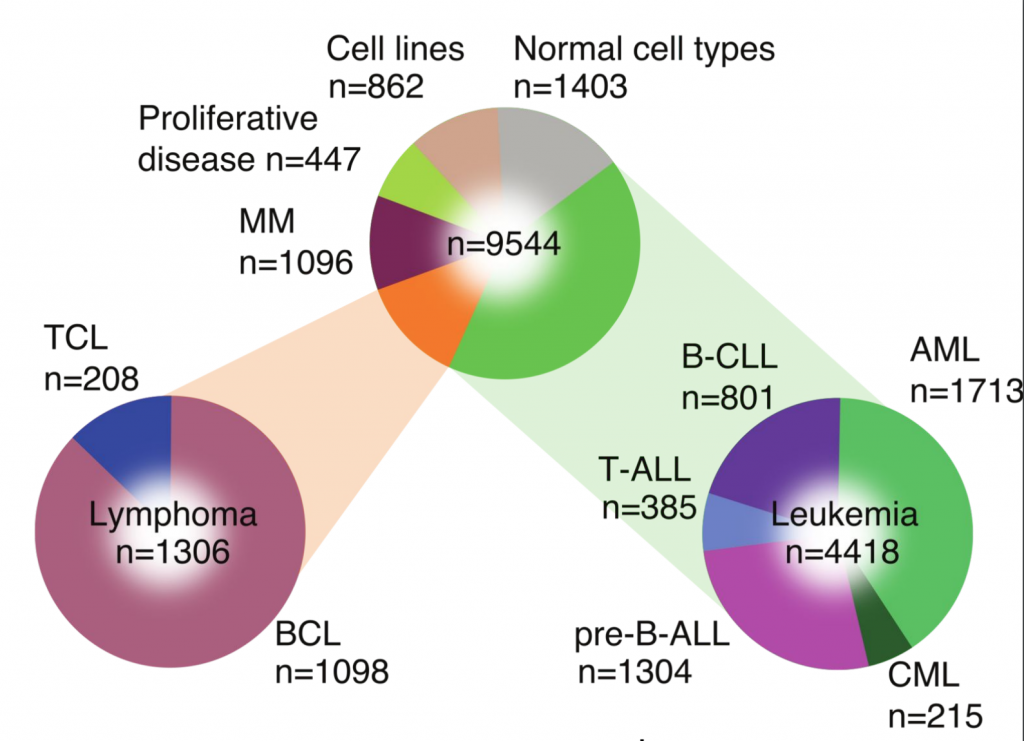
 We manually curated and analyzed 9,544 transcriptomes from more than 30 hematologic malignancies, normal blood cell types, and cell lines, and showed that disease types could be stratified in a data-driven manner. We identified cluster-specific pathway activity, new biomarkers and in silico drug target prioritization through interrogation of drug target databases. Using known vulnerabilities and available drug screens, we highlighted the importance of integrating molecular phenotype with drug target expression for in silico prediction of drug responsiveness. These molecular data can be explored using our publicly available interactive resource, Hemap, for expediting therapeutic innovations in hematologic malignancies.
We manually curated and analyzed 9,544 transcriptomes from more than 30 hematologic malignancies, normal blood cell types, and cell lines, and showed that disease types could be stratified in a data-driven manner. We identified cluster-specific pathway activity, new biomarkers and in silico drug target prioritization through interrogation of drug target databases. Using known vulnerabilities and available drug screens, we highlighted the importance of integrating molecular phenotype with drug target expression for in silico prediction of drug responsiveness. These molecular data can be explored using our publicly available interactive resource, Hemap, for expediting therapeutic innovations in hematologic malignancies.
Computational analysis of immune microenvironments in glioblastoma
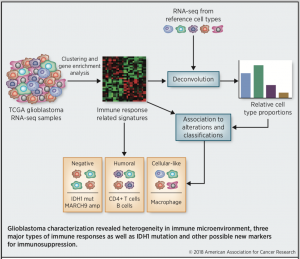
Glioblastoma is the most common malignant brain tumor in adults, and patient prognosis remains poor with current therapies. While the anti-tumor immune response is actively suppressed in glioblastoma microenvironment, modern immunotherapies have generated promising treatment responses in a subpopulation of glioblastoma patients. However, an understanding of the nature of immunosuppression is still largely lacking although it is important for successful cancer treatment through immune system modulation in a personalized fashion. To answer this need, we performed a computational analysis to model the relative immune cell content and type of immune response in glioblastoma tumor samples from TCGA RNA-seq dataset. Our analysis identified three major subgroups of microenvironments in TCGA glioblastoma cohort (Negative, Humoral, Cellular-like) presenting different prevailing immune response. These subgroups and immune cell compositions were associated with typical genetic alterations and transcriptional GBM subtypes. Overall, our analysis revealed high heterogeneity in the immune microenvironment of GBM and identified possible new markers for immunosuppression.
Tissue Reconstruction Algorithms for 3D Histology
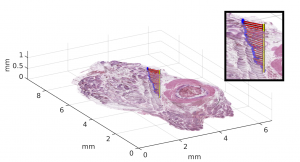
Digital pathology enables 3D histology, where a three-dimensional reconstruction of the sample is formed computationally based on serial tissue sections. This allows examining tissue architecture in 3D, and joint mapping of cellular morphology with spatially resolved omics data in the true 3D context of the tissue at microscopic resolution. We developed a benchmarking framework to evaluate the accuracy of several free and commercial 3D reconstruction methods using two whole slide image datasets. The results provide a solid basis for further development and application of 3D histology algorithms.
Integrative proteogenomics of prostate cancer
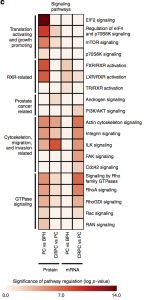
We report high-throughput mass spectrometry with integrated genome level analysis on clinical tissue samples of benign prostatic hyperplasia (BPH), untreated primary prostate cancer (PC) and castration resistant prostate cancer (CRPC). Each sample group shows a distinct protein profile. We show that, especially in CRPC, gene copy number, DNA methylation, and RNA expression levels do not reliably predict proteomic changes. Instead, we uncover previously unrecognized molecular and pathway events, for example, several miRNA target correlations present at protein but not at mRNA level. Notably, we identify two metabolic shifts in the citric acid cycle (TCA cycle) during prostate cancer development and progression.
See Research highlight article “Controlling Mutational Chaos” in Nature Reviews Urology.
Circulating tumor DNA genomics correlate with drug resistance in prostate cancer
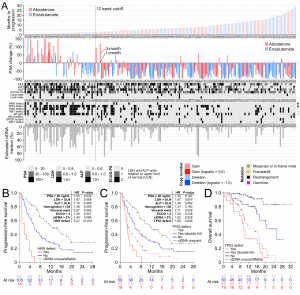
In collaboration with Dr. Alexander Wyatt and team from Vancouver Prostate Centre, we have studied primary resistance to androgen receptor (AR) directed therapies in metastatic castration-resistant prostate cancer (mCRPC). We randomized 202 treatment-naive mCRPC patients to abiraterone or enzalutamide, and performed whole exome and deep targeted 72-gene sequencing of plasma cell-free DNA prior to therapy. For these agents, which have never been directly compared, time to progression was similar. Our analysis shows that defects in BRCA2 and ATM were strongly associated with poor clinical outcomes independently of clinical prognostic factors and circulating tumor DNA abundance. Somatic alterations in TP53, previously linked to reduced tumor dependency on AR signaling, were also independently associated with rapid resistance. Although detection of AR amplifications did not outperform standard prognostic biomarkers, AR gene structural rearrangements truncating the ligand binding domain were identified in several patients with primary resistance. These findings establish genomic drivers of resistance to first-line AR directed therapy in mCRPC and identify potential minimally-invasive biomarkers.
Strong FGFR3 staining is a marker for FGFR3 fusions in diffuse gliomas
 We have previously identified and characterized FGFR3 fusions in glioblastoma. Here we performed FGFR3 immunohistochemistry on tissue microarrays containing 676 grades II–IV astrocytomas and 116 grades II–III oligodendroglial tumor specimens. Fifty-one cases were further analyzed using targeted sequencing. Moderate to strong FGFR3 staining was detected in gliomas of all grades, was more common in females, and was associated with poor survival in diffuse astrocytomas. Strong FGFR3 protein expression is indicative of FGFR3 fusions and may serve as a clinically applicable predictive marker for treatment regimens based on FGFR inhibitors.
We have previously identified and characterized FGFR3 fusions in glioblastoma. Here we performed FGFR3 immunohistochemistry on tissue microarrays containing 676 grades II–IV astrocytomas and 116 grades II–III oligodendroglial tumor specimens. Fifty-one cases were further analyzed using targeted sequencing. Moderate to strong FGFR3 staining was detected in gliomas of all grades, was more common in females, and was associated with poor survival in diffuse astrocytomas. Strong FGFR3 protein expression is indicative of FGFR3 fusions and may serve as a clinically applicable predictive marker for treatment regimens based on FGFR inhibitors.
Decision support for pathologists: machine learning based image interpretation
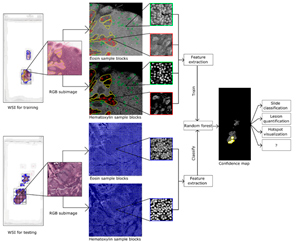
Digital pathology has led to a demand for automated detection of regions of interest, such as cancerous tissue, from scanned whole slide images. Our study shows how machine learning based image interpretation can be used for detecting the cancer hot spots from WSIs. Our method was able to detect metastatic tissue from breast cancer lymph nodes samples with high accuracy (AUC in tumor tissue detection ranging between 0.84-0.91). Based on random forest classification model, we were also able to find a shortlist of top contributing features, providing insight into differences in spatial properties of the tissue types.
DNA repair gene loss is detectable in circulating tumor DNA
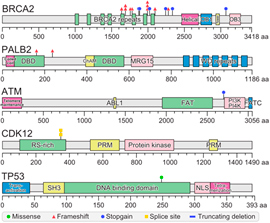
Germline mutations in DNA repair genes have been recently reported in 8 – 12% of patients with metastatic castration resistant prostate cancer (mCRPC). In this study, we performed targeted germline sequencing of 319 metastatic castration resistant prostate cancer patients and showed that patients carrying deleterious germline mutations in genes linked to homologous recombination exhibit attenuated responses to androgen receptor targeted therapy. Using cell-free DNA sequencing, we showed that germline BRCA2 mutations associated with somatic loss-of-heterozygosity in all but one case. These results suggest that mCRPC patients with germline DNA repair defects can be prioritized for more effective therapies using a blood test.